

従来の遺伝子発現解析
= 細胞集団をまとめて解析
➔ 全体の平均が分かる

scRNA-seq
= 個々の細胞を解析
➔ 個々の細胞の違いが分かる
組織を構成する個々の細胞の遺伝子発現を調べる方法です。
Bulk RNA-Seqでは細胞集団における平均的な遺伝子発現を検出するのに対して、個々の細胞の遺伝子発現を調べる single cell RNA-seq 解析(scRNA-seq)により、組織を構成する様々な細胞の機能・活性化状態が詳細に解析できます。
・
BD Rhapsody システムにおいてナノウェル・磁気ビーズを利用した whole transcriptome scRNA-seq 解析です。
*日本ベクトン・ディッキンソン株式会社よりBD Rhapsody™ シングルセル解析システムを使用したサービスプロバイダーとして認定されております
・
当社独自の固相ビーズからの全cDNA増幅法 (TAS-Seq法) により、高感度・高精度な解析を実現しています。
・
マルチプレックス解析に対応しており、多くのコストを必要とせず多検体解析が可能です。(USB法により細胞種・生物種によらず標識可能です。 『技術紹介』のマルチプレックス項目をご参照ください)
・
mRNAと細胞表面タンパク質の発現量を同時に測定する(CITE-seq : Cellular Indexing of Transcriptomes and Epitopes by sequencing)も実施可能です。『お問い合わせ』よりご連絡ください。
・
BD社 SampleTag、BioLegend社 Hashtag等によるマルチプレックス解析に対応しており、多くのコストを必要とせず多検体解析が可能です。
・
BD Rhapsody Express システムの貸し出し、現地へ作業員の派遣も可能です。 (別途費用)
・
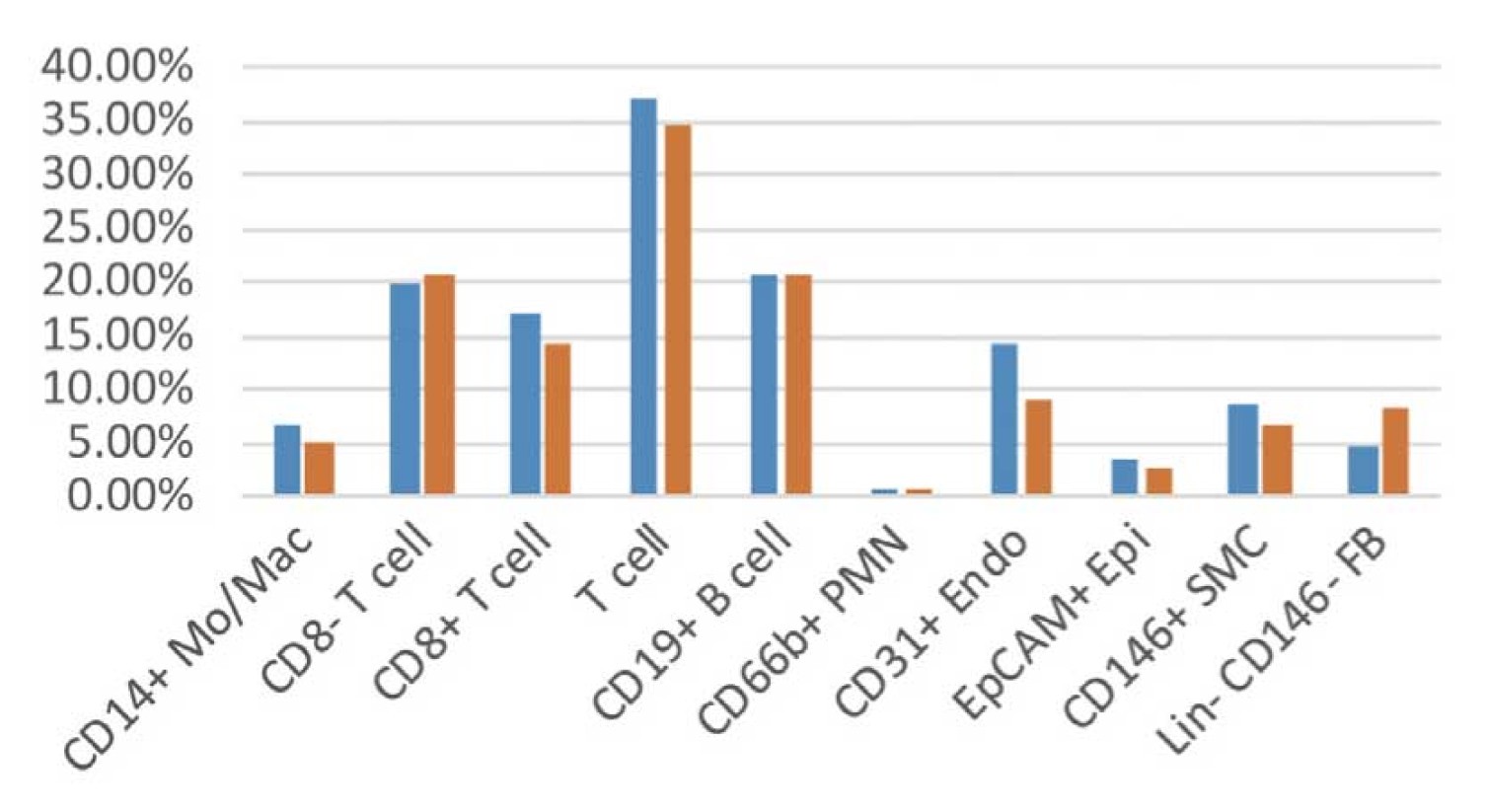
マウス肺、すい臓ラ氏島、腎臓や、ヒト肺、胃がんの生検サンプル由来白血球、ヒト末梢血好中球などの解析実績があります。
・
解析利用論文はこちら
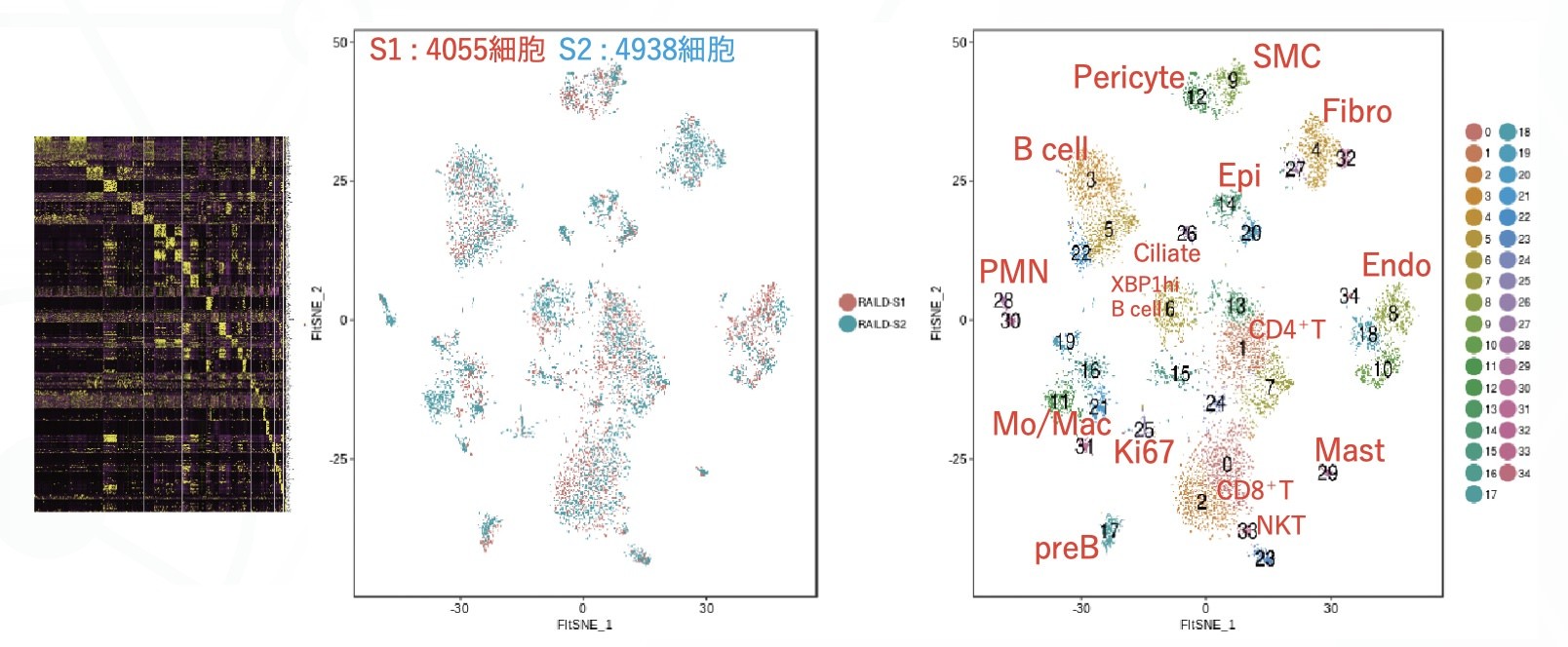
ヒトリウマチ肺の解析例
S1:4055細胞、S2:4938細胞をプールし、PCA空間上でクラスタリング後、その結果をtSNEによる次元圧縮により可視化しました。
ImmunoGeneTeqsのRhapsody とTAS-Seqを組み合わせた手法では、Batch effectがほとんど無く、基本的にはBatch effect補正の必要がありません。
* クラスタリングとは似たような遺伝子発現パターンを持った細胞を探してクラスター(集団)に分けることを指します。
* 異なる実験環境(Batch)で計測されたデータの場合、同じ臓器・細胞であっても、Batch間でデータに差が生じる" Batch effect "(実験間誤差)が存在し、それらの影響によりクラスタリング結果の解釈など、データ解釈が困難となる場合、Batch effect補正が必要な場合もありますが、Batch effect補正を実施することでbiological な違いもマスクしてしまう可能性があります。
マウス肺細胞におけるフローサイトメーターと各種手法の細胞検出精度の比較
フローサイトメーターのデータと比較してTAS-seqではフローサイトメーターとの相関が高く、正確な細胞組成データを取得可能です。
その他の手法の10X v2データはマクロファージを過大に検出しており、線維芽細胞画分を過小に検出していました。 (赤枠を参照)
Smart-seq2 データは内皮細胞と単球を過大に検出しており、肺胞マクロファージが失われていました。 (緑枠を参照)
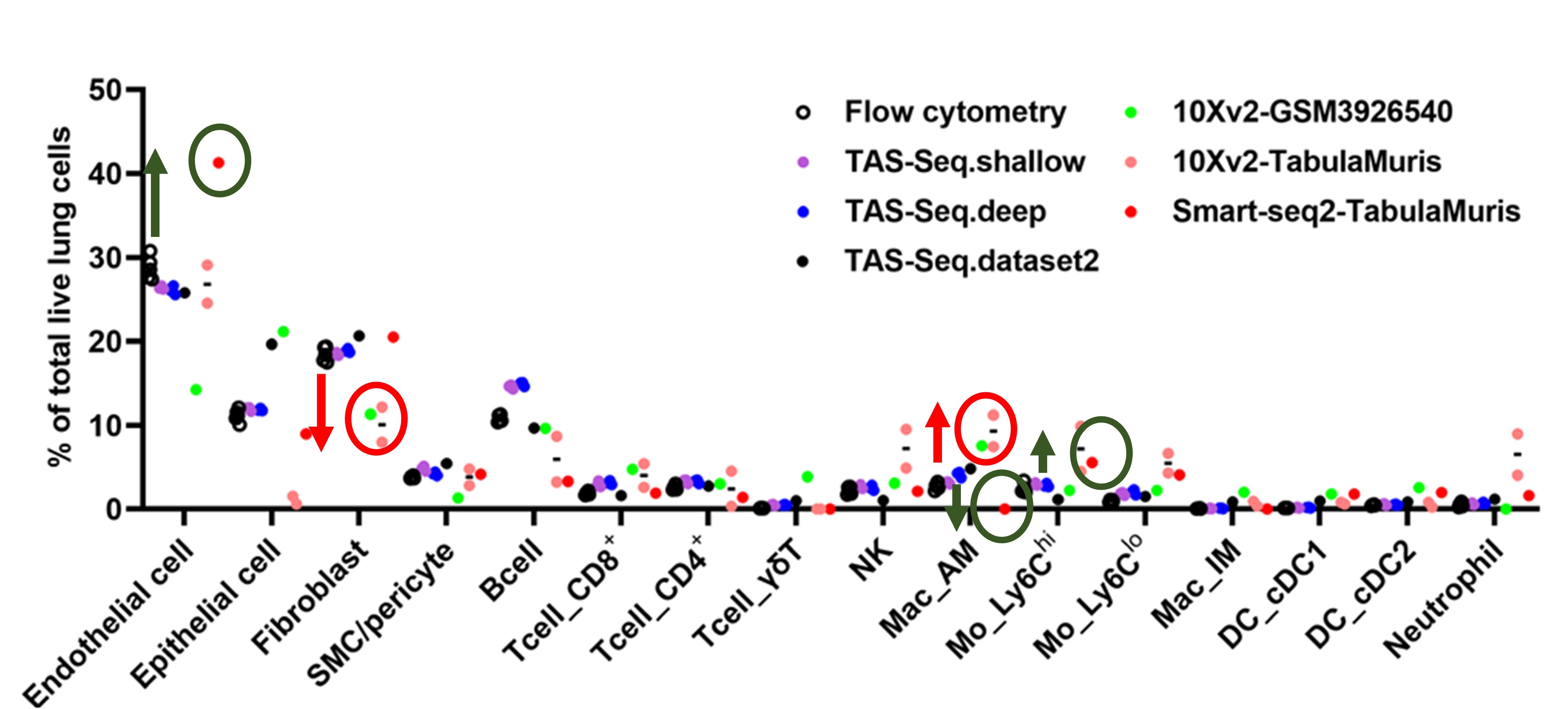



Shichino S. et al. Communications Biology volume 5, Article number: 602 (2022)
マウス肺の細胞間コミュニケーション検出




TAS-seq2の開発
TAS-Seqの反応系を最適化し、検出感度がさらに向上したTAS-Seq2を開発致しました。


TAS-Seq2を用いたマウス肝臓検体由来細胞核解析例

*10X 核調整キット、anti-nuclear pore complex hashtagsを利用 約20000-30000 reads / nucleus
TAS-Seq2により、細胞核の測定においても他社手法と比較して高感度に遺伝子を検出

10X v3.1: 公開済みマウス肝臓検体データ(SRX14774301, SRX14774300)
解析実績のあるサンプル
single-cell RNA-seq
マウス
等
ヒト
等
培養細胞
等
その他生物種
等
single-nucleus RNA-seq
マウス
等

必要な機材・試薬を
弊社よりお客様へ送付

➊ 細胞のロード
(自然落下・ポワソン分布)
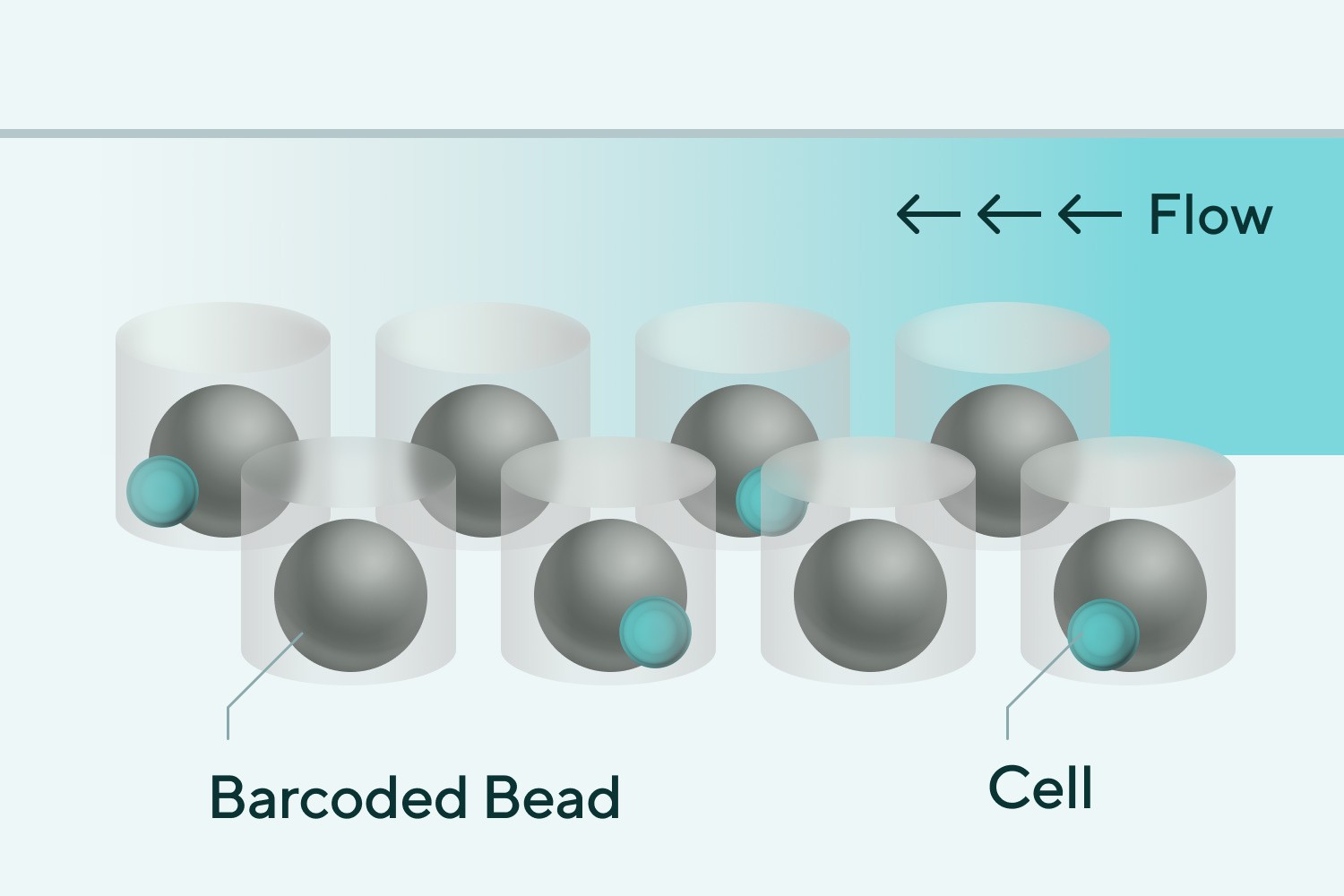
➋ 過剰量のビーズのロード
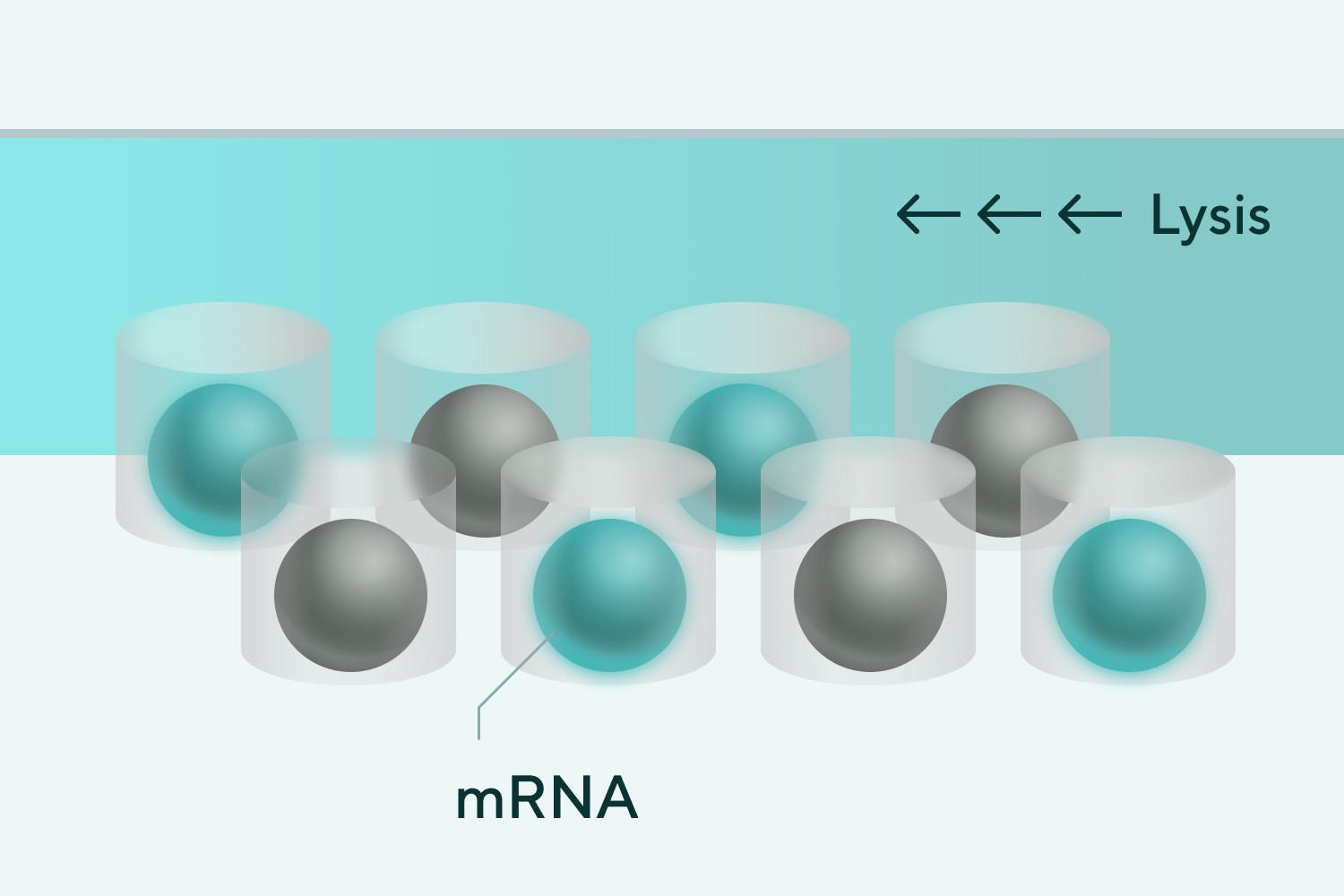
➌ 細胞溶液、各種細胞由来mRNAをビーズに補足
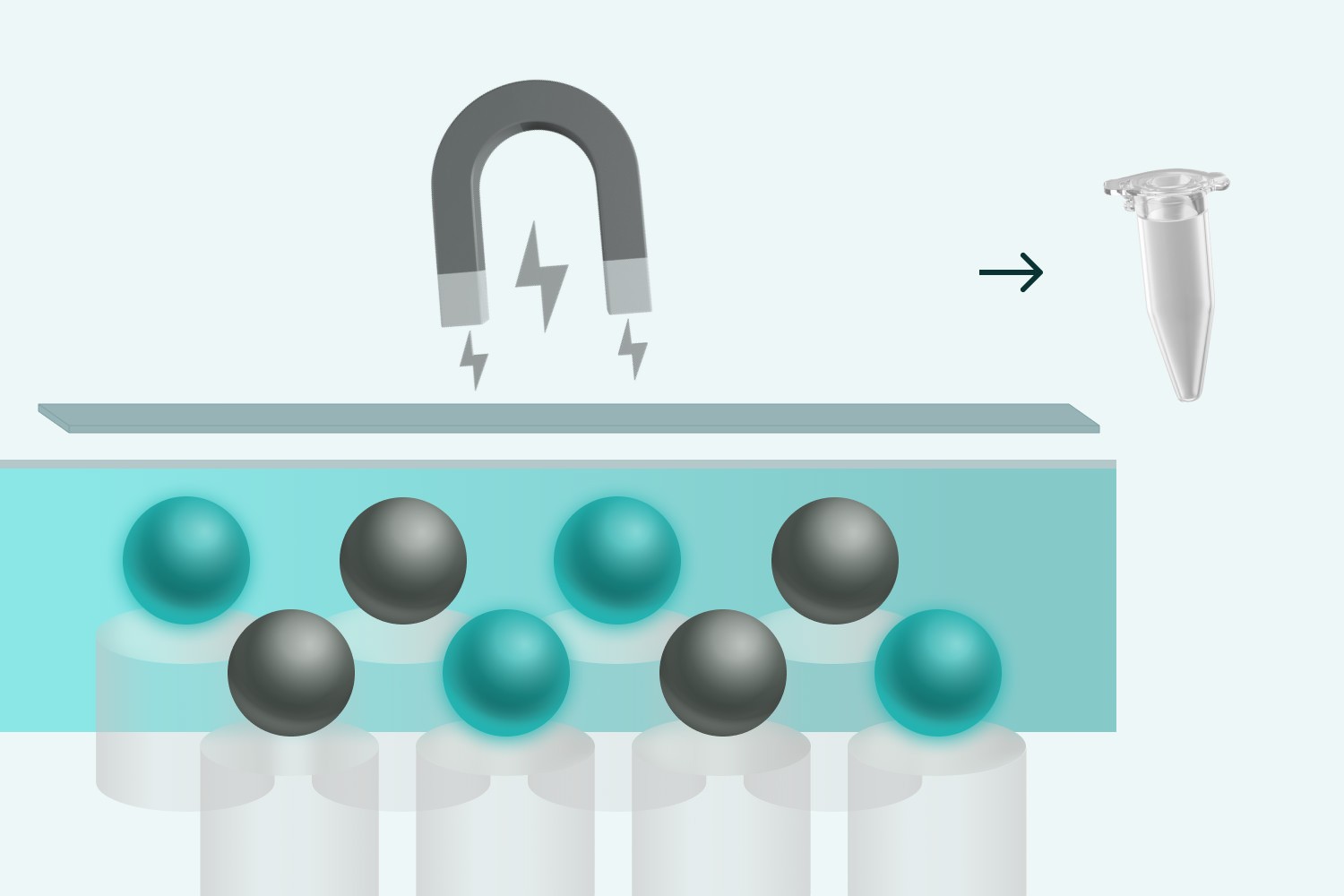
➍ ビーズの回収・逆転写

お客様がオンサイトにて➊~➍まで行ったサンプルを当社へ送付
事前確認事項
* マルチプレックス解析をご希望の際は、FAQページの『マルチプレックス解析について』をご参照ください。
ご依頼方法
サンプルの受け入れ
・
当社指定プロトコルにて合成頂いたcDNA (IGT社でcDNA増幅以降を受託)
・
凍結細胞(IGT社で細胞抗体染色やcDNA合成以降を受託)
・
凍結組織(IGT社で組織からの細胞懸濁液調整以降を受託)
* 凍結細胞、組織から調整をご希望の場合はお問合わせください
* 細胞や組織を凍結保存頂く際は、当社指定の保存液、凍結方法をご使用下さい
サンプル送付方法
・
サンプルは乾燥・液漏れが無いようご注意いただき、下記発送先までご送付ください (平日17時到着まで対応可能)
・
当社指定プロトコルにて逆転写済みのBD Rhapsody beadsを発送する場合は、冷蔵便にてご発送ください
・
凍結細胞・組織をご発送の場合は、凍結状態を維持するために十分量のドライアイスを同梱し冷凍便でお送りください
* 遠隔地を除き翌日必着でご送付ください
サンプル送付先
〒277-0882
千葉県柏市柏の葉6丁目6番2号
三井リンクラボ柏の葉1 304号室
TEL:04-7192-8732
イムノジェネテクス株式会社
納期
サンプル受領から
2.5ヶ月程度
納品物 (HDD)
・
作業報告書
・
シーケンス生データ一式
・
マッピング結果ファイル(遺伝子発現テーブル並びにRNA velocity解析用発現テーブル、解析レポートファイル等)
シーケンスデータから1細胞ごとの遺伝子発現テーブルを作成いたします
報告書を抜粋したもの をご確認いただけます
報告書のうちSeurat解析のレポートはオプションとなり、別途費用をいただきます
報告書の例としてオプション解析も含めた場合を、Latest Dataからご覧いただけます
追加解析
Seurat ソフトウェアによるクラスター解析やマーカー遺伝子テーブルの作成も承っております(別途費用)
参考価格
1
20,000
2
20,000
4
10,000
8

Analysis Report
マッピング解析レポート
マッピング解析の概略
1.
得られたシーケンスから、アダプター配列を除去します。
2.
品質の高い配列を取得し、サイクルごとの塩基組成を確認し、シーケンシングに問題がないことを確認します。
3.
得られたシーケンスのcDNA部分について、参照配列にマッピングします。
4.
マッピングされたシーケンスデータを、各1細胞に対応する固有のバーコード配列に基づき分割し、各細胞の遺伝子発現データを得ます。
5.
Barcode rank plot(図1)に基づき、分析に有効な細胞を検出します。またscatter plotにより、得られた細胞ごとのリード数と検出遺伝子数の分布につき確認することができます。
結果例
・
表1では、cDNA部分のマッピング率、シーケンスリードの利用効率、最終的に得られた細胞数、細胞集団の平均遺伝子検出数などについての情報を示します。
・
図1にBarcode rank plotを示します。これは有効な細胞数を決定するためのプロットです。
表1 マッピングの統計:検出された細胞と遺伝子の数
項目
数または割合 (%)
図1 Barcode rank plot
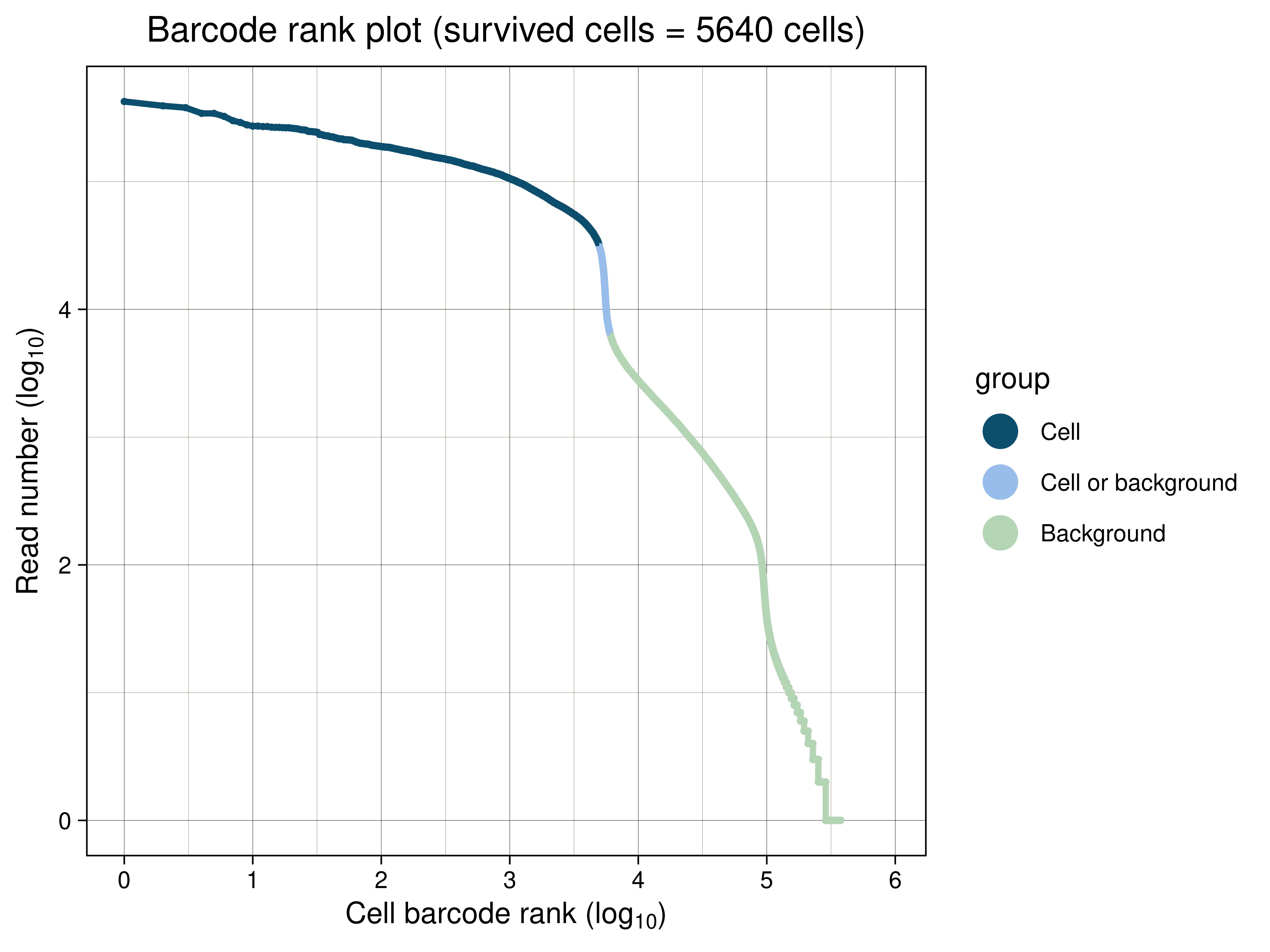
縦軸は細胞バーコードごとのリード数。横軸は細胞バーコードあたりのリード数の多い順に各細胞バーコードに付けられた順位。変曲点がマゼンタの縦線で示されている。
1) データの前処理
・
tag (BioLegend社Hashtag、BD社Sampletagなど) を用いて1度に複数サンプルを同時解析する場合、各細胞のtag発現量を求め、各細胞におけるtag発現量の多寡に基づいて、どの細胞がどのサンプル由来であるのかを決定 (図2、表2)
表2 各サンプルタグに割り当てられた細胞の数
図2
図2 各細胞バーコードのサンプルタグ発現量と、各細胞にアサインされたtagの関係をヒートマップで図示

2) Seuratにおける前処理
・
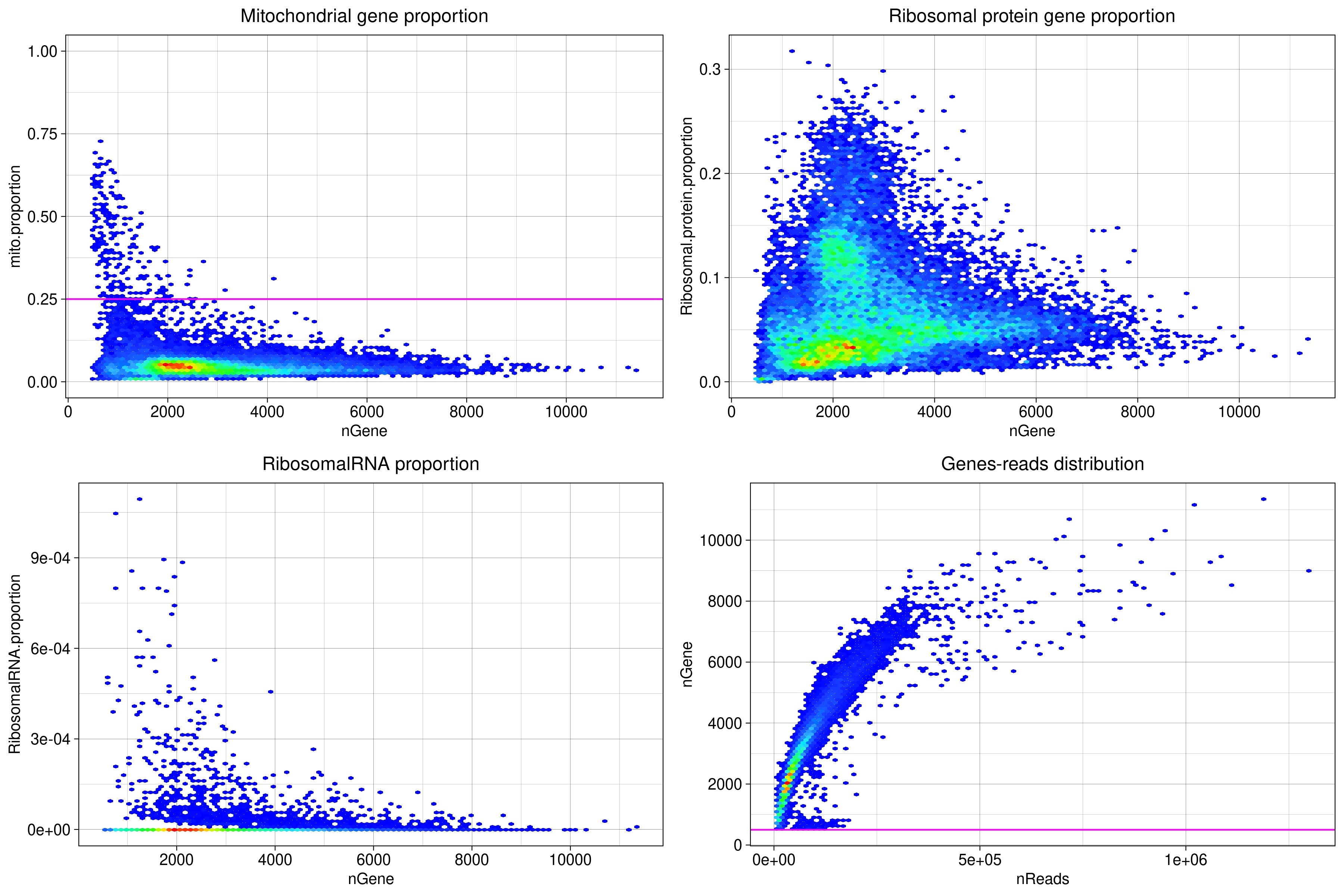
ミトコンドリア遺伝子の発現が高い細胞 (0.25以上) の除去 (図3)
・
ダブレット細胞およびタグが発現していなかった細胞を除去 (tagを用いた解析の場合)
・
ScaleData関数を使用したスケーリング
・
Pseudocolor density plotやRidgeplotにより、検出遺伝子数および遺伝子あたりの発現数の分布を求め、問題がないか概ね確認
図3 統計データのpseudocolor 密度プロット
この図においては、1つの点が1つの細胞に対応し、細胞が同じ位置で重なるところはその重なりの密度を求め、密度が低い方から濃い方へ青から赤のスペクトルに対応させて、その密度に応じたスペクトル内の色で表されています。
3) 主成分分析とJackStrawプロット
大きく発現変動している遺伝子をFindVariableFeatures関数で検出した後、それら遺伝子に関する主成分分析(PCA)を実施します。遺伝子発現パターンに基づく細胞のクラスタリングのために使用する主成分(PC)の数を、Jackstraw解析により決定します
図4 JackStrawプロット
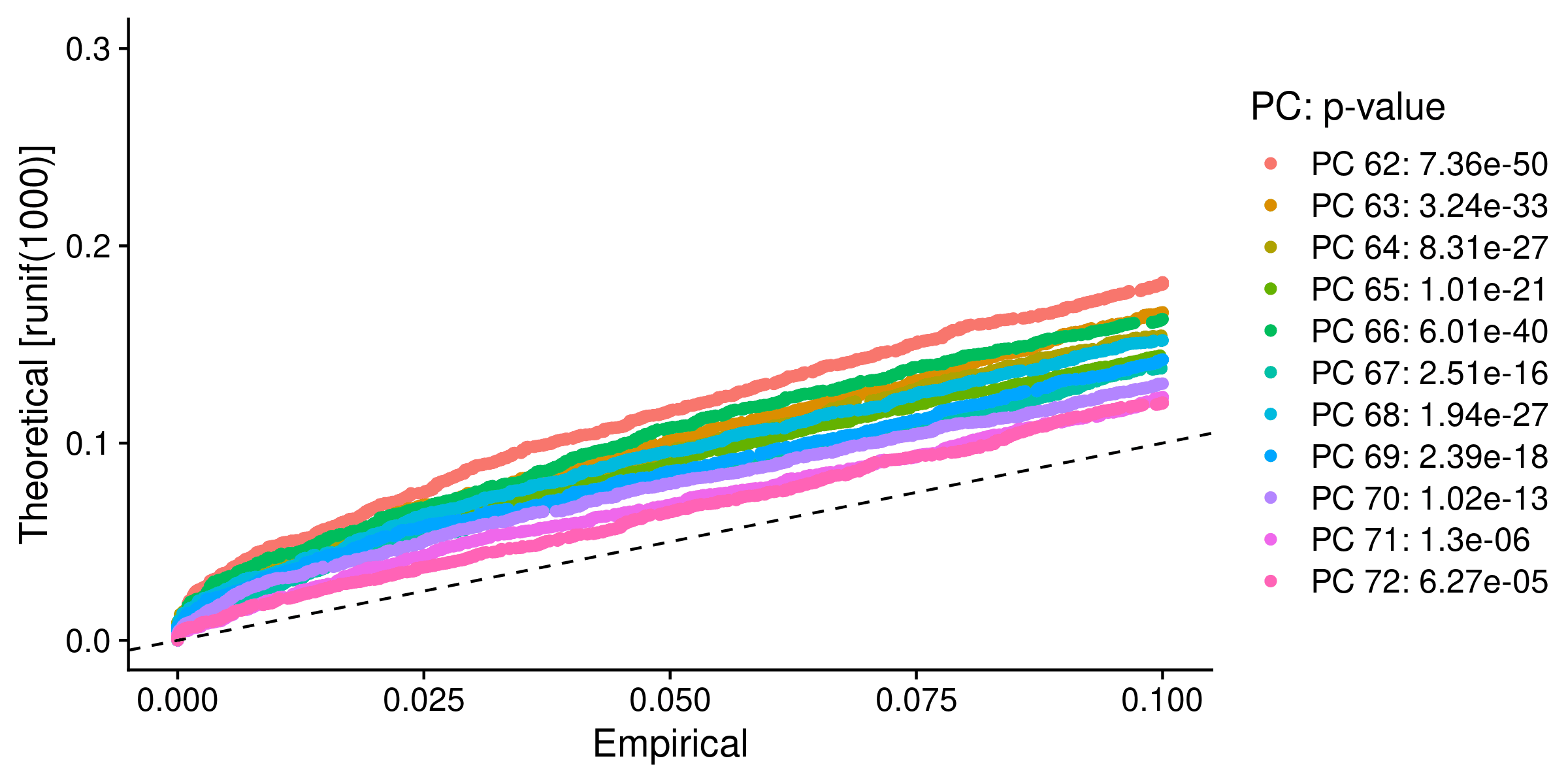
p値が1 x 10-5未満となる71までの主成分数を、以降の解析に使うことにしました。
4) クラスタリング
細胞のクラスタリングを実行します。遺伝子発現の似た細胞が1つのクラスターとなります。
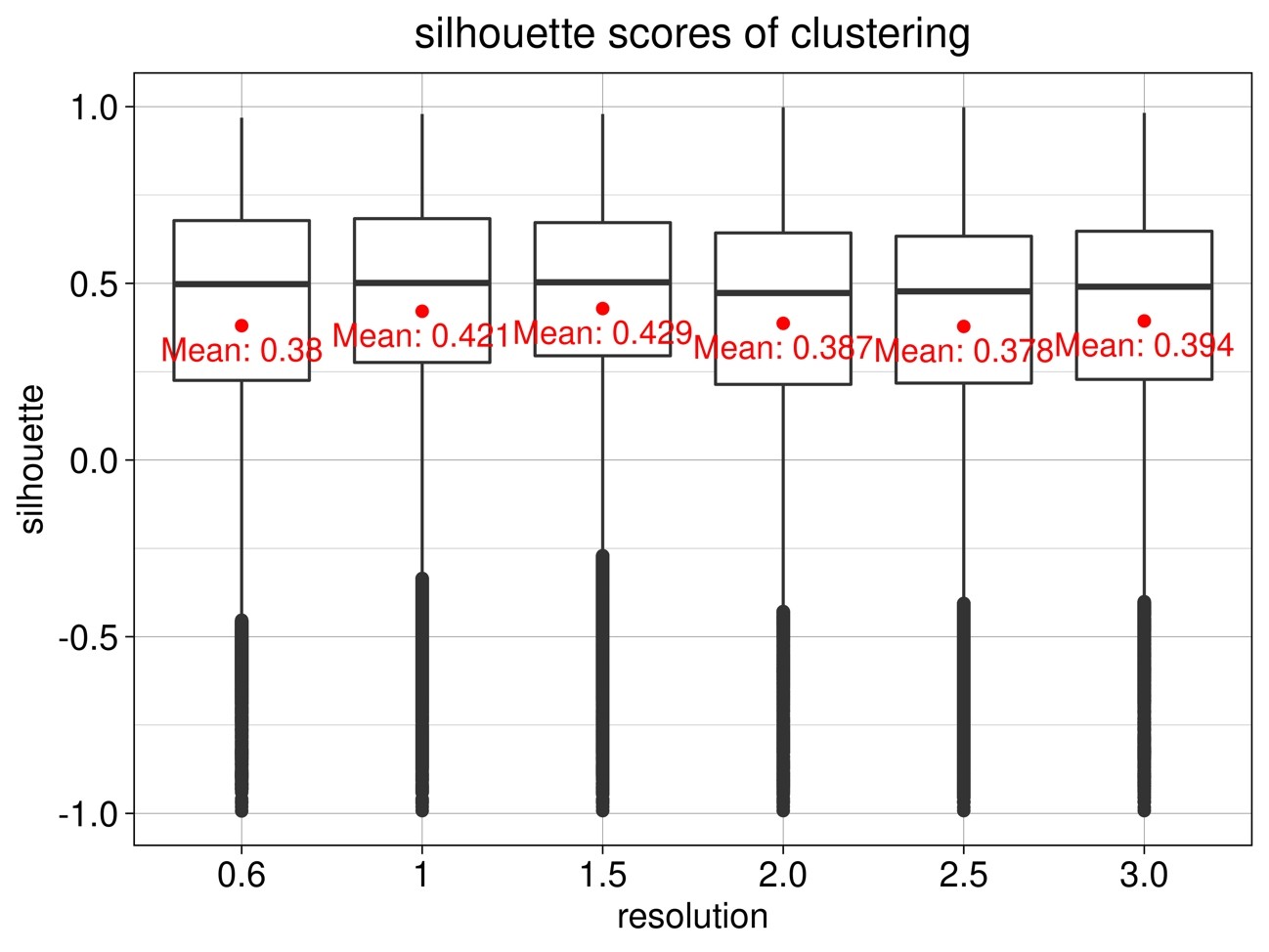
5) Seuratクラスタリングシルエットスコアプロット
解像度パラメータを決定します。 (図5) ここでは、解像度1.5において平均シルエットスコアが最大なので、1.5が適切と考えて以降の解析に進んでいます。
図5 それぞれの解像度パラメータにおけるクラスタリングの平均シルエットスコア
6) Fit-SNEプロットおよびUMAPプロットによるSeuratクラスタリング結果の可視化
FIt-SNE (George C. Linderman et al. Nat Methods 2019) を用いて、4)で行ったクラスタリング結果を視覚化します 。(図6) またUMAP解析でも視覚化します (図7)
図6 クラスタリング結果のFlt-SNEによる表示
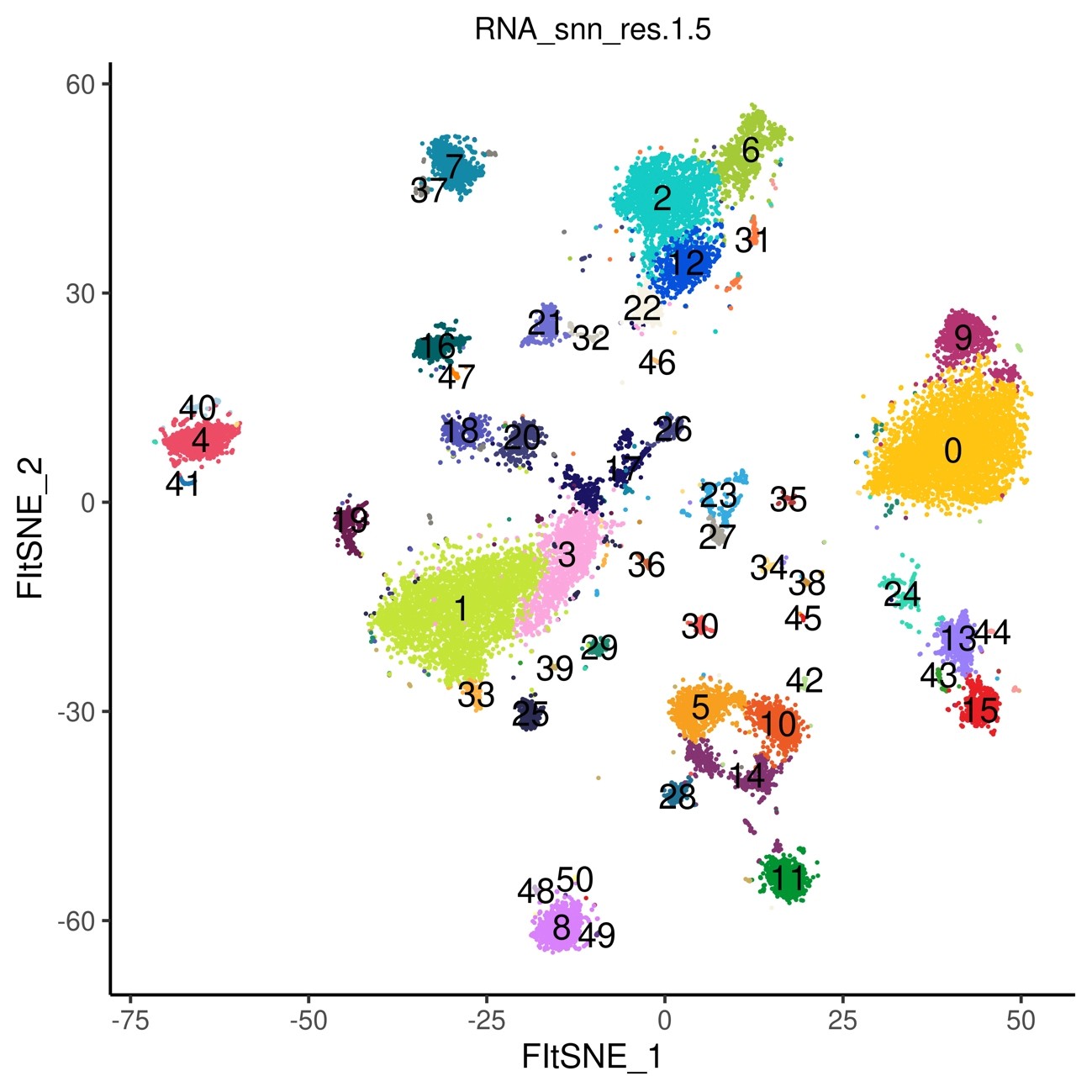
50個のクラスターが検出されました。
図7 クラスタリング結果のUMAPによる表示
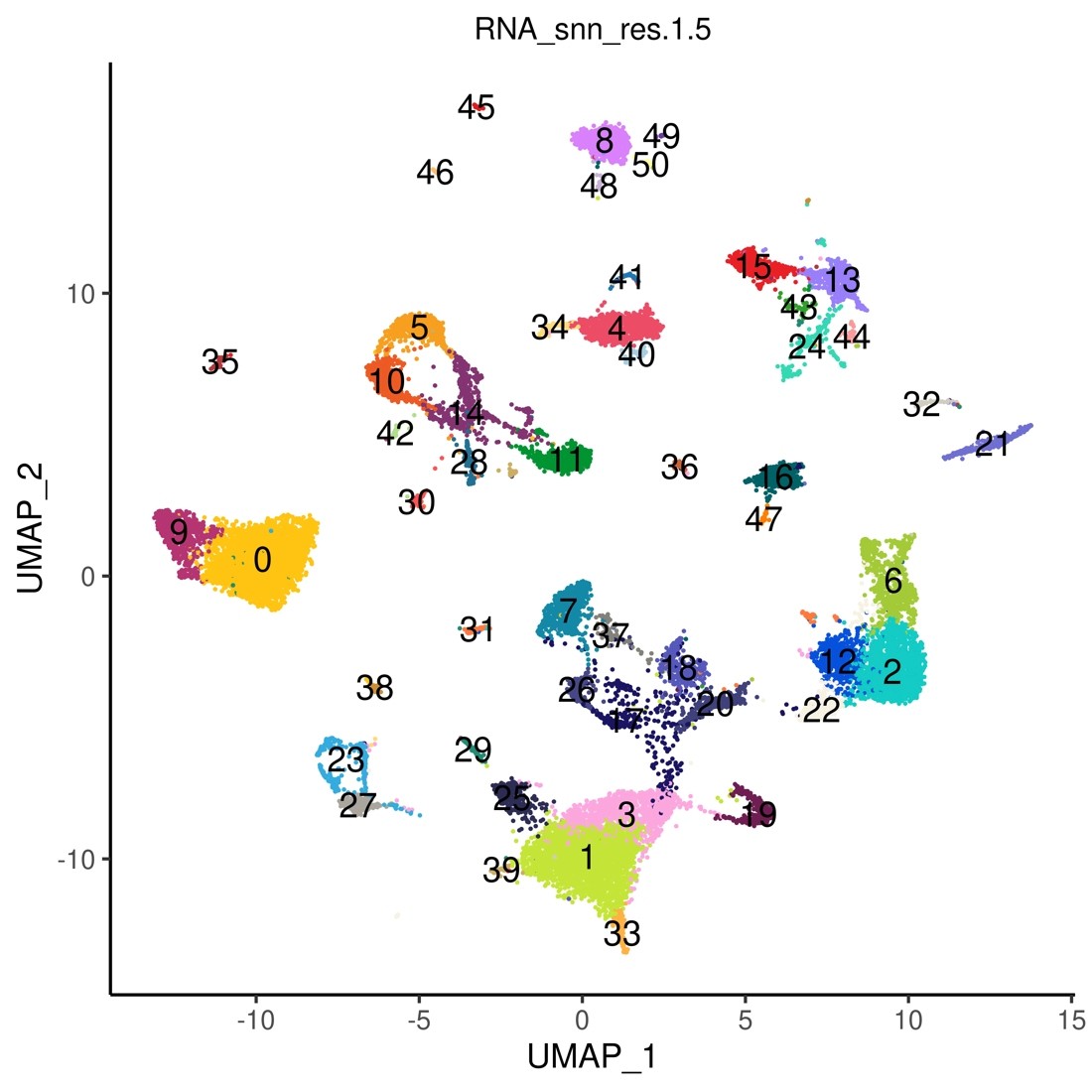
50個のクラスターが検出されました。
7) 各サンプルにおける細胞組成の解析
・
各クラスターに含まれる細胞数を求めます (表3)
・
サンプル間の細胞組成の違いを求めます (図8、図9)
表3 タグごと(サンプルごと)の各クラスターに含まれる細胞数
0
529
785
558
526
587
1
672
640
458
352
651
2
284
272
334
273
338
3
228
243
198
172
208
4
182
150
167
135
176
5
114
232
164
138
120
6
171
130
150
158
149
7
167
148
100
213
113
8
113
210
159
110
123
9
121
146
140
110
157
・
クラスター番号は、図6、図7のSueratクラスター番号に該当します。0から9までのみ表示しています。
・
元のデータは.txtに保存されます。またhtmlファイルでも全体を表示することができます。
図8
各細胞クラスターの全細胞に対する割合を、サンプルごとに積み上げ棒グラフで図示
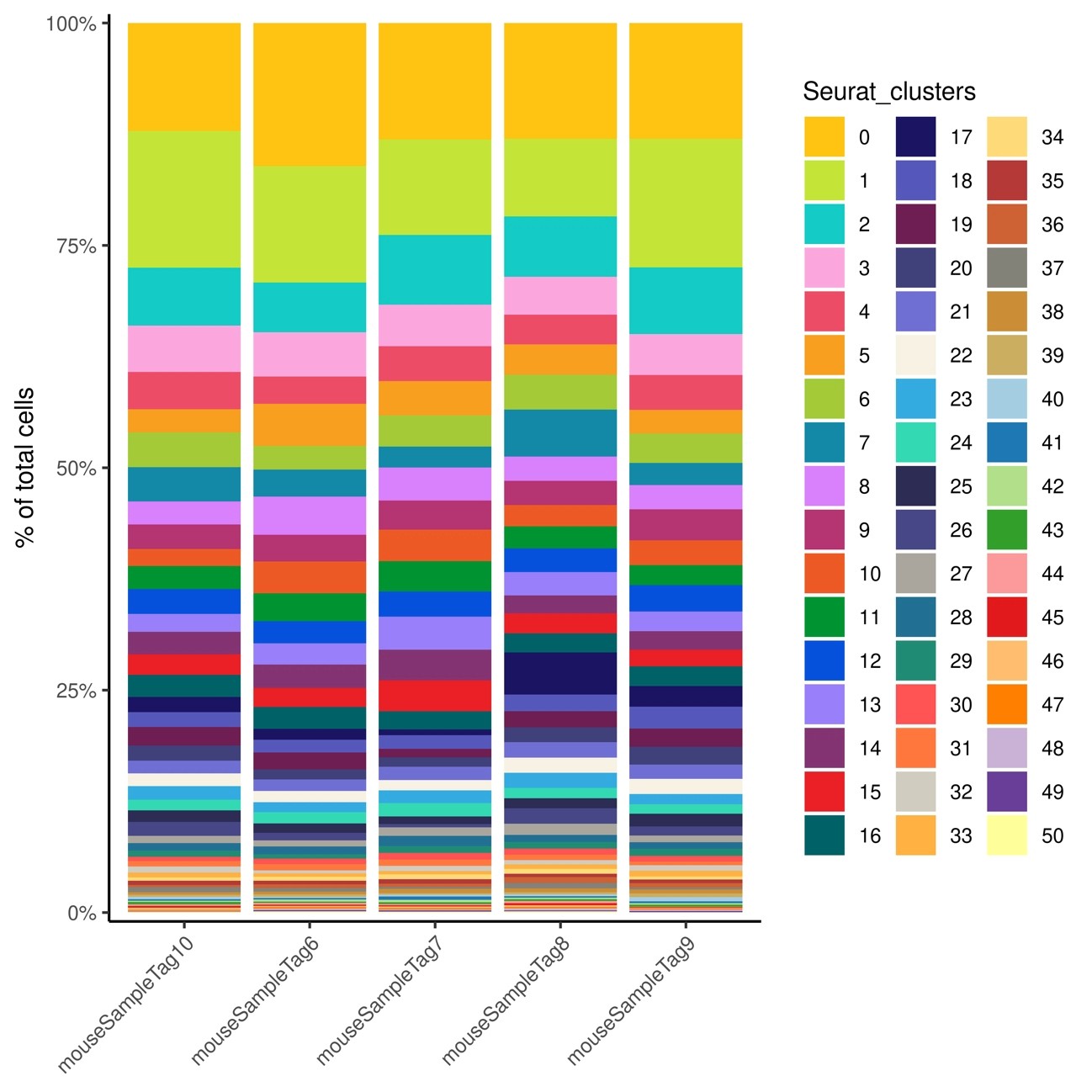
50個のクラスターが検出されました。
図9
細胞組成割合のサンプル間での差異に関する系統樹解析
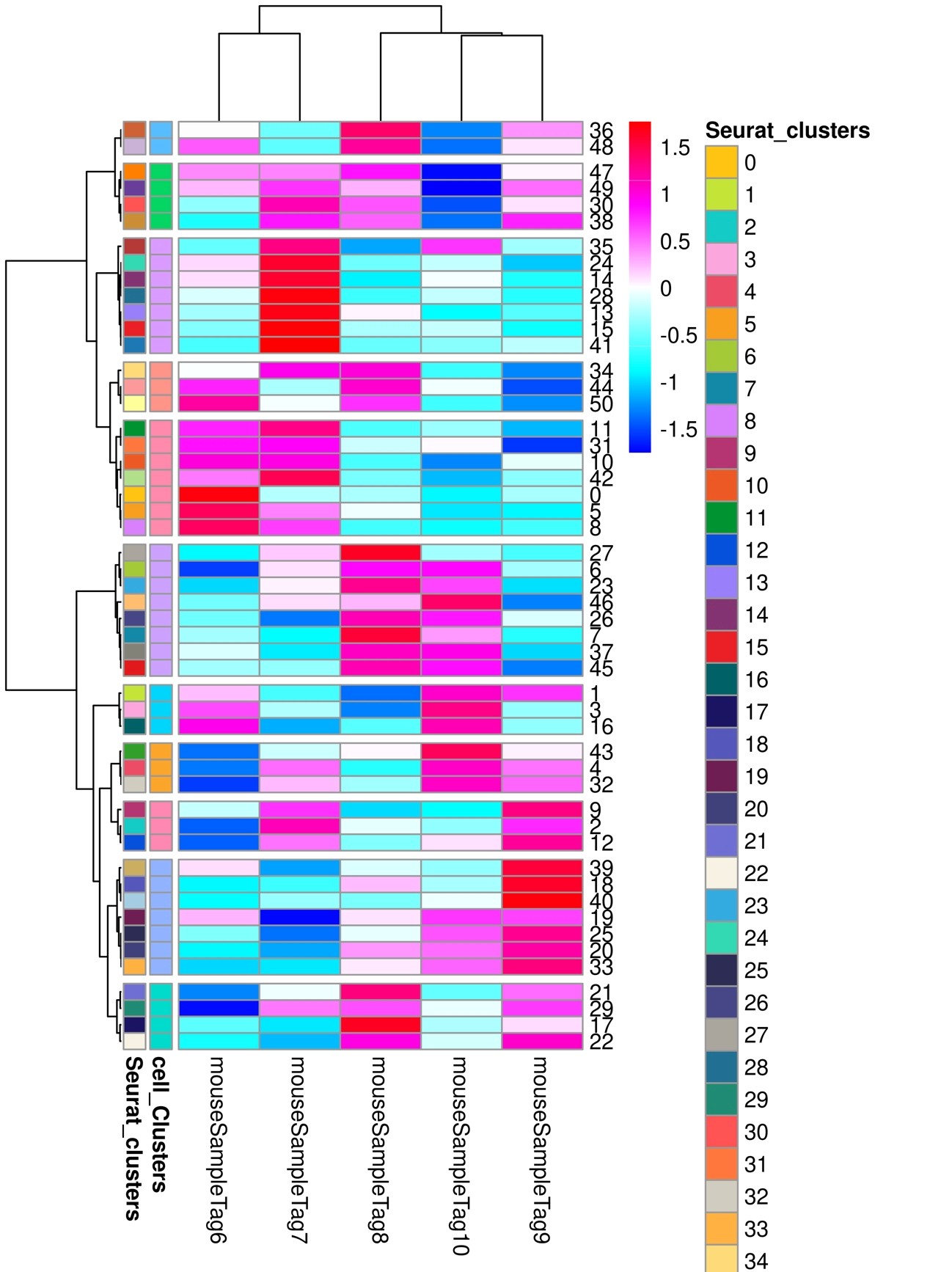
50個のクラスターが検出されました。
8) 各細胞クラスターにおけるマーカー遺伝子の検出
・
各細胞クラスターにおいて特徴的に高く発現しているマーカー遺伝子を見つけます (図10、表4)
・
トップ6のマーカー遺伝子の発現パターンをFIt-SNE、UMAPにより可視化しました(図11、図12)
図10 各クラスターにおけるトップ20のマーカー遺伝子の発現パターンをヒートマップで図示
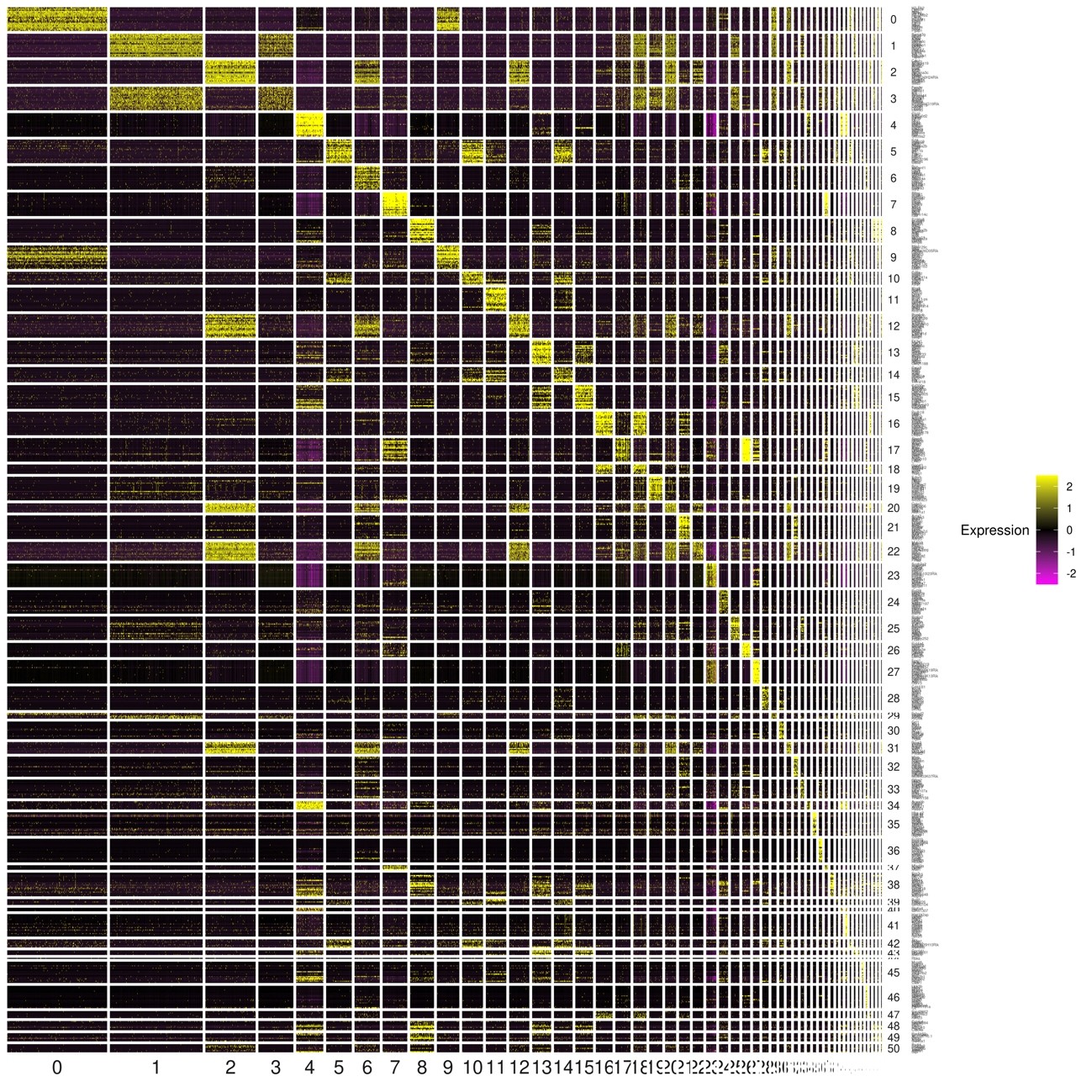
縦軸に各クラスターにおいて特徴的な20の遺伝子、横軸には各クラスターにある個々の細胞を並べ、個々の細胞における遺伝子発現量を右にあるスペクトルの色で表しました。クラスター内にある個々の細胞の遺伝子発現がどのくらい類似度があるか、あるいはクラスターごとの遺伝子発現の比較などができます。
表4 各クラスターにおいて検出された全マーカー遺伝子
gene: クラスターにおけるマーカー遺伝子
cluster: Seuratクラスター
p_val_adj: 変動遺伝子検定の結果におけるbonferroni補正p値
logFC: あるクラスターと、それ以外のすべてのクラスターにおける平均発現量のlog2単位で表した変化
within_avg_exp: あるクラスターにおける平均発現量(ln)
without_avg_exp: あるクラスター以外のすべてのクラスターでの平均発現量(ln)
pct.1: あるクラスターにおいて発現している細胞の割合
pct.2: 該当クラスター以外のすべてにおいて発現している細胞の割合
p_val: 変動遺伝子検定結果のp値
*マーカー遺伝子テーブルは.txtに保存されます。またhtmlファイルでも遺伝子名やクラスター名を検索したり、値を並べ替えたりすることができます。
図11 各クラスターにおけるトップ6のマーカー遺伝子の発現パターン (Flt-SNE)
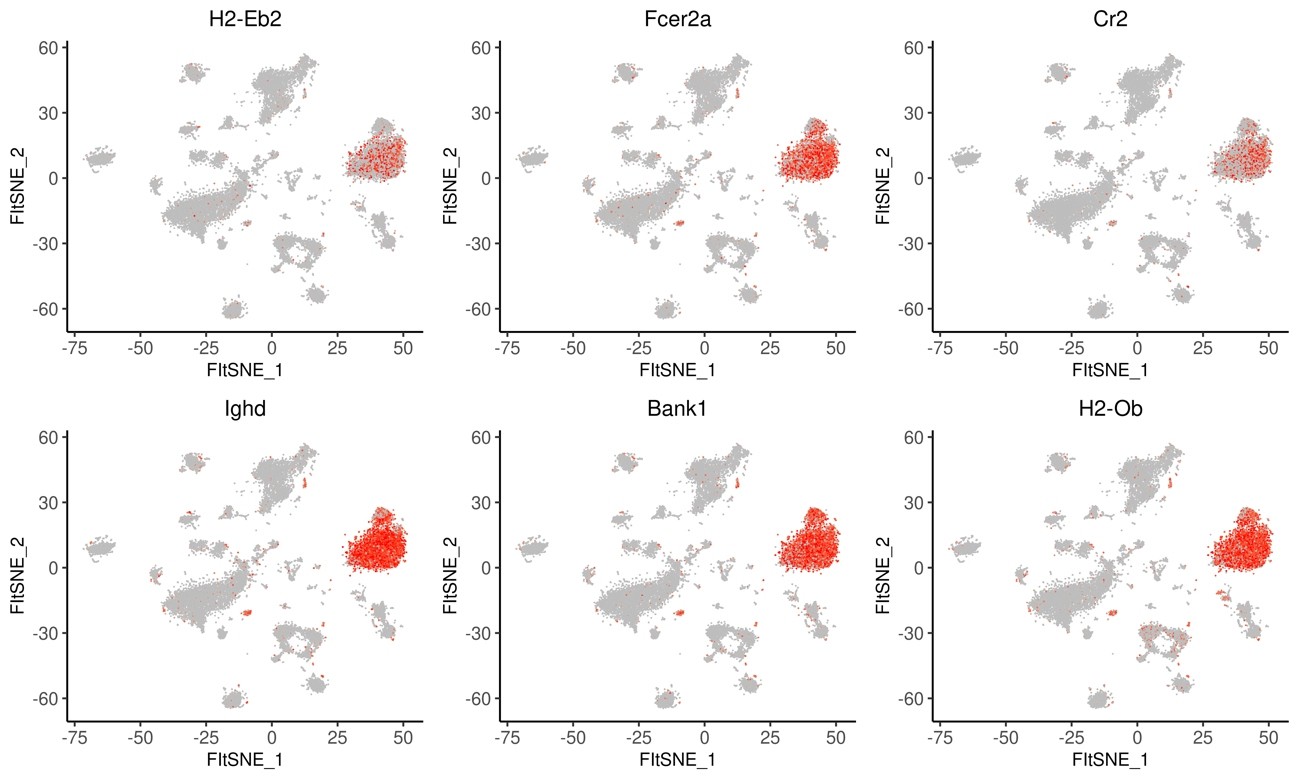
図6で示されたクラスターのうち、クラスター0についての各遺伝子の発現量を示しました。htmlファイルではそれぞれのクラスターにつき表示を切り替えることができます。
図12 各クラスターにおけるトップ6のマーカー遺伝子の発現パターン (UMAP)
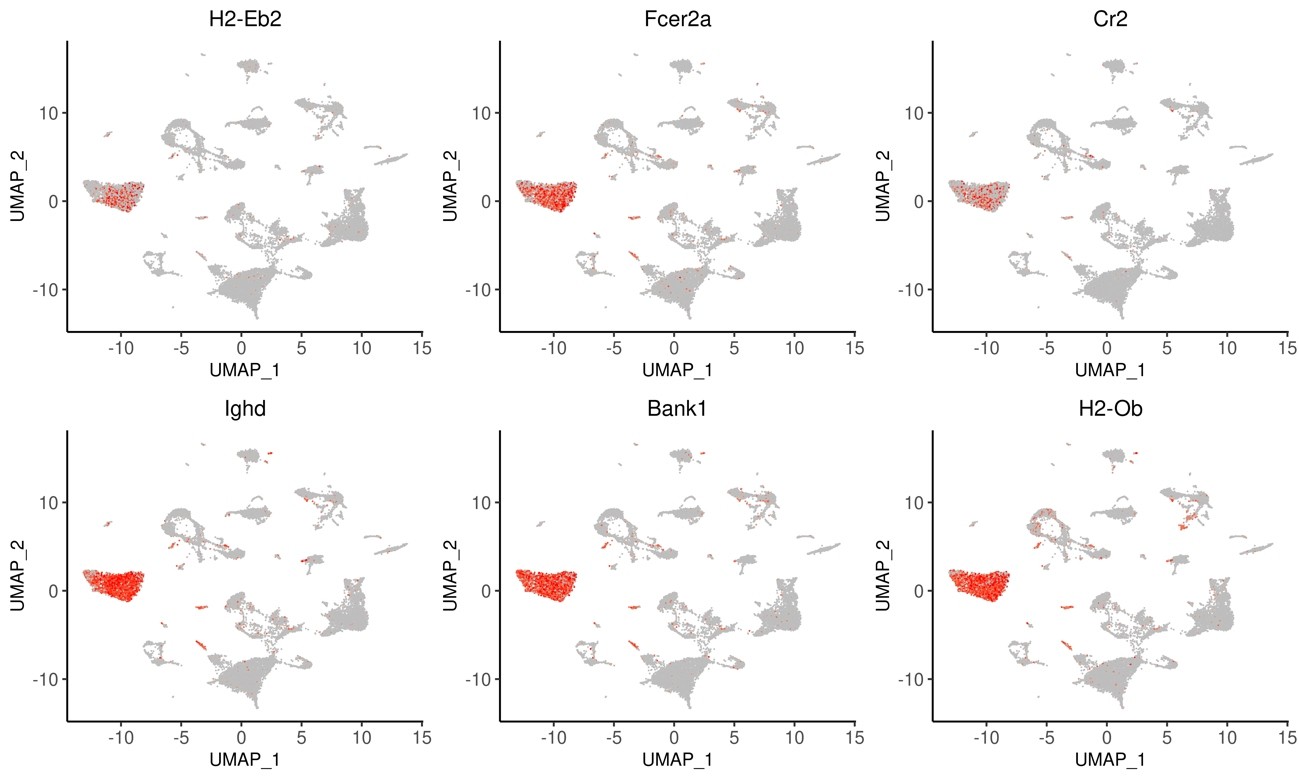
図7で示されたクラスターのうち、クラスター0についての各遺伝子の発現量を示しました。htmlファイルではそれぞれのクラスターにつき表示を切り替えることができます。
9) 細胞種の推定 (参考データ)
各クラスターに含まれる個々の細胞の種類につき、SingleRパッケージにより推定します。(図13、図14) あくまでも参考データですので、検出されたマーカー遺伝子とも突き合わせつつ、ご自身で各クラスターが何の細胞なのかをマニュアルでアノテーションし、確認されることをお勧めいたします。
図13 各クラスター中の推定された細胞種の割合
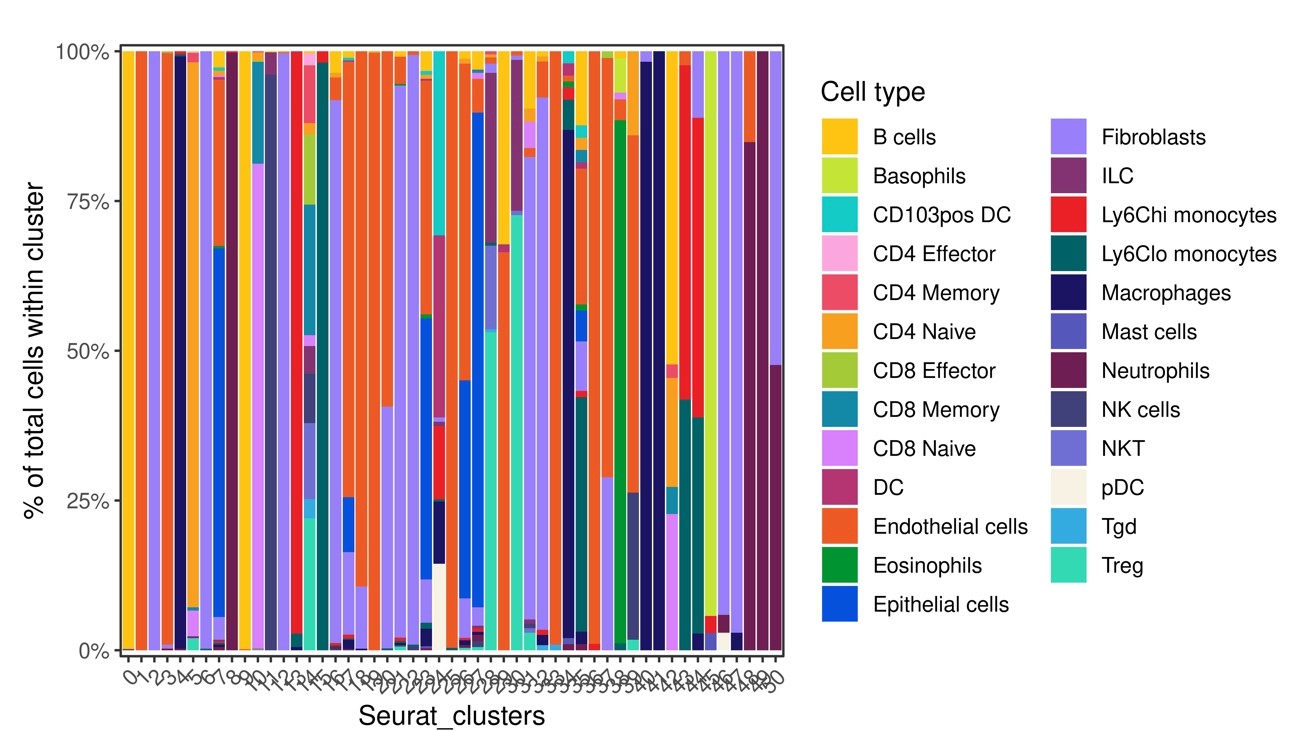
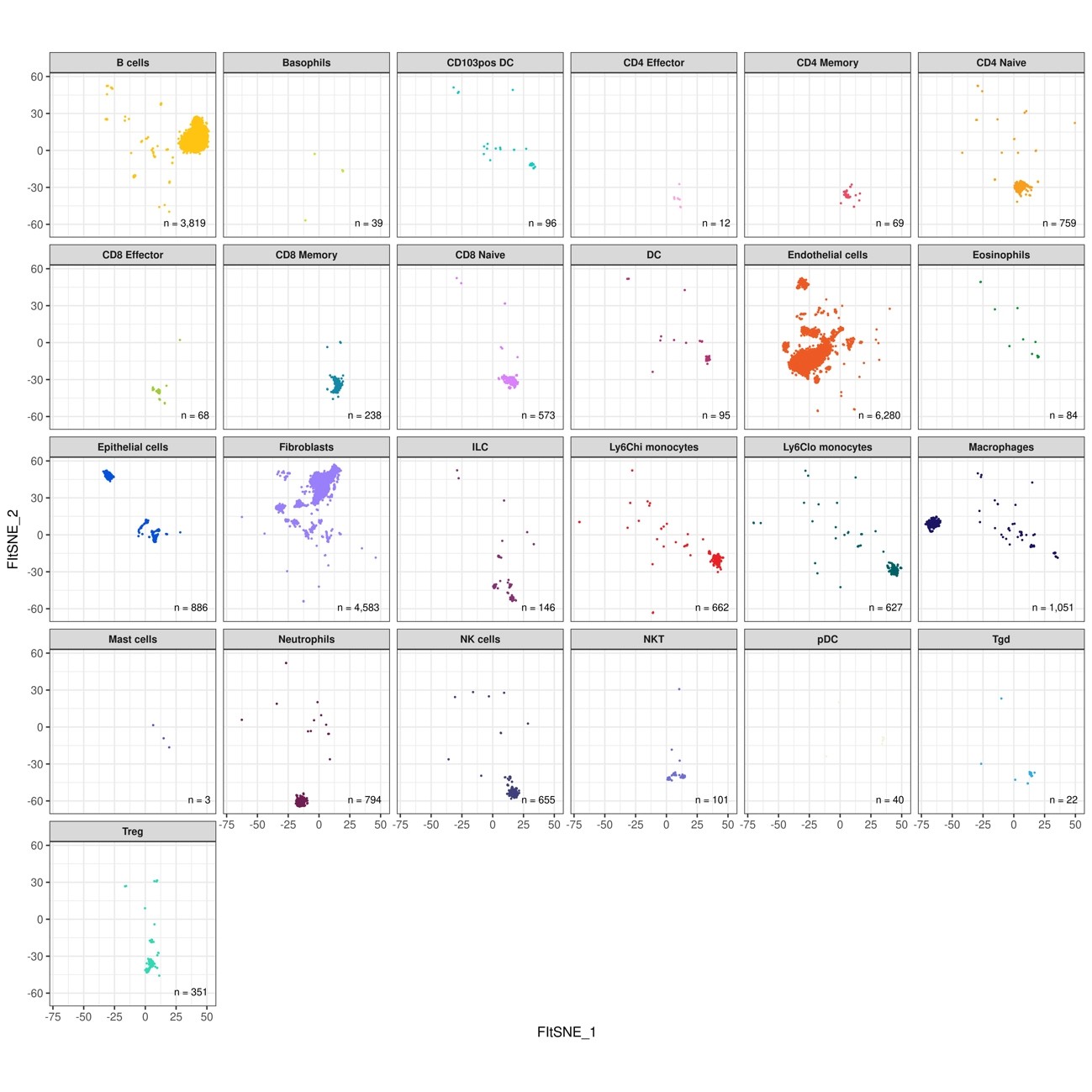
図14 推定された各細胞種のFIt-SNE空間上の分布を表示

TAS-Seq is a robust and sensitive amplification method for bead-based scRNA-seq
Human iPS cell-derived cartilaginous tissue spatially and functionally replaces nucleus pulposus
Combining an Alarmin HMGN1 Peptide with PD-L1 Blockade Results in Robust Antitumor Effects with a Concomitant Increase of Stem-Like/Progenitor Exhausted CD8+ T Cells
AMPK activation reverts mouse epiblast stem cells to naive state
Single cell transcriptomics clarifies the basophil differentiation trajectory and identifies pre-basophils upstream of mature basophils
Angiopoietin-like 4 is a critical regulator of fibroblasts during pulmonary fibrosis development
The early neutrophil-committed progenitors aberrantly differentiate into immunoregulatory monocytes during emergency myelopoiesis